

Posttransplant lymphoproliferative disorders (PTLD) are a spectrum of disorders characterized by lymphoid or plasmacytic proliferation secondary to extrinsic immunosuppression in solid organ transplant (SOT) or hematopoietic transplant recipients. According the 2016 revision of World Health Organization (WHO) classification, PTLD ranges from early lesion (mononucleosis-like) lymphoid hyperplasia or plasmacytic hyperplasia, polymorphic, monomorphic to classical Hodgkin lymphoma PTLD (Swerdlow et. al. Blood 2016). The incidence of PTLD in the pediatric population is closely associated with the type of transplanted organ, with the highest incidence in lung and small bowel transplant recipients due to chronic, iatrogenic immunosuppression. While Epstein-Barr Virus (EBV) is the key driver of abnormal lymphocytic proliferation in the majority of PTLDs, EBV-negative PTLD is a distinct subtype.
We hereby report a 9-year-old boy with Barth syndrome, a rare genetic syndrome caused by a TAZ mutation, characterized by dilated cardiomyopathy, skeletal myopathy, neutropenia, and short stature. He required a heart transplant for left ventricular non-compaction cardiomyopathy at 11 months of age and presented with lower GI bleeding, acute-onset anemia, somnolence, retinal hemorrhages, and increased serum viscosity eight years post transplant. Workup was significant for hyperimmunoglobulinemia with elevated IgA and IgG, and serum protein electrophoresis (SPEP) showed two monoclonal bands (IgA Lambda and IgG Kappa) and an M spike. He was treated with plasmapheresis, and his symptoms improved significantly. Further workup with PET/CT scan showed diffuse lymphadenopathy of cervical, mediastinal, axillary, abdominal, and inguinal lymph nodes. Axillary lymph node biopsy was done, demonstrating a nondestructive morphology with an increase in plasma cells expressing excess lambda light chain and IgA; these abnormal cells were CD20 negative and CD138 positive, suggesting a clonal process. Cytogenetic fluorescence in situ hybridization (FISH) showed loss of TP53. Serum EBV detection by PCR and EBER staining by immunohistochemistry (IHC) on lymph node pathology were negative. Bone marrow biopsy showed a hypocellular marrow with trilineage hematopoiesis and 5-10% CD138 positive plasma cells. Combined, these studies were consistent with the diagnosis of early onset, nondestructive, plasmacytic PTLD.
The patient was treated with a multiple myeloma-type therapy with dexamethasone and bortezomib (Short et. al. J Pediatr Hematol Oncol 2016), in addition to reduced immunosuppression consisting of low dose tacrolimus. Repeat PET/CT scan after 2 cycles of therapy showed resolution of previous PET-avid lesions. Stem cell collection was performed after 3 cycles in case of relapsed or recurrent disease. He completed 5 cycles of therapy with no complications.
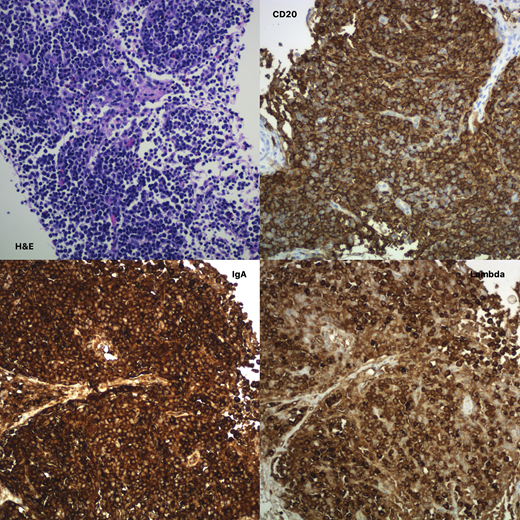
Unfortunately, the patient's end of treatment PET/CT scan was significant for a new mediastinal mass and multiple intraabdominal lymph nodes. Lymph node biopsy showed a lymphoplasmacytic infiltrate, which was IgA and lambda positive. The cells showed plasmacytoid features, but in contrast to the initial biopsy at diagnosis, there was marked CD20 positivity (image attached). This recurrence was most suggestive of a marginal zone lymphoma like PTLD (Galera et. al. Am J Surg Pathol 2020). He was initiated on therapy with rituximab and bendamustine. He is currently status post 2 treatment cycles and doing well. We continue to discuss the utility of autologous bone marrow transplant in this case, although the risks and benefits will have to be weighed carefully in a child who is prone to develop infections and complications from Barth Syndrome.
We would like to share this case of EBV negative, plasmacytic PTLD with relapsed marginal zone lymphoma type PTLD to highlight the heterozygous pathologies of PTLD in children, and to share our experience of treating in the absence of standard protocol due to the rarity of this diagnosis.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal